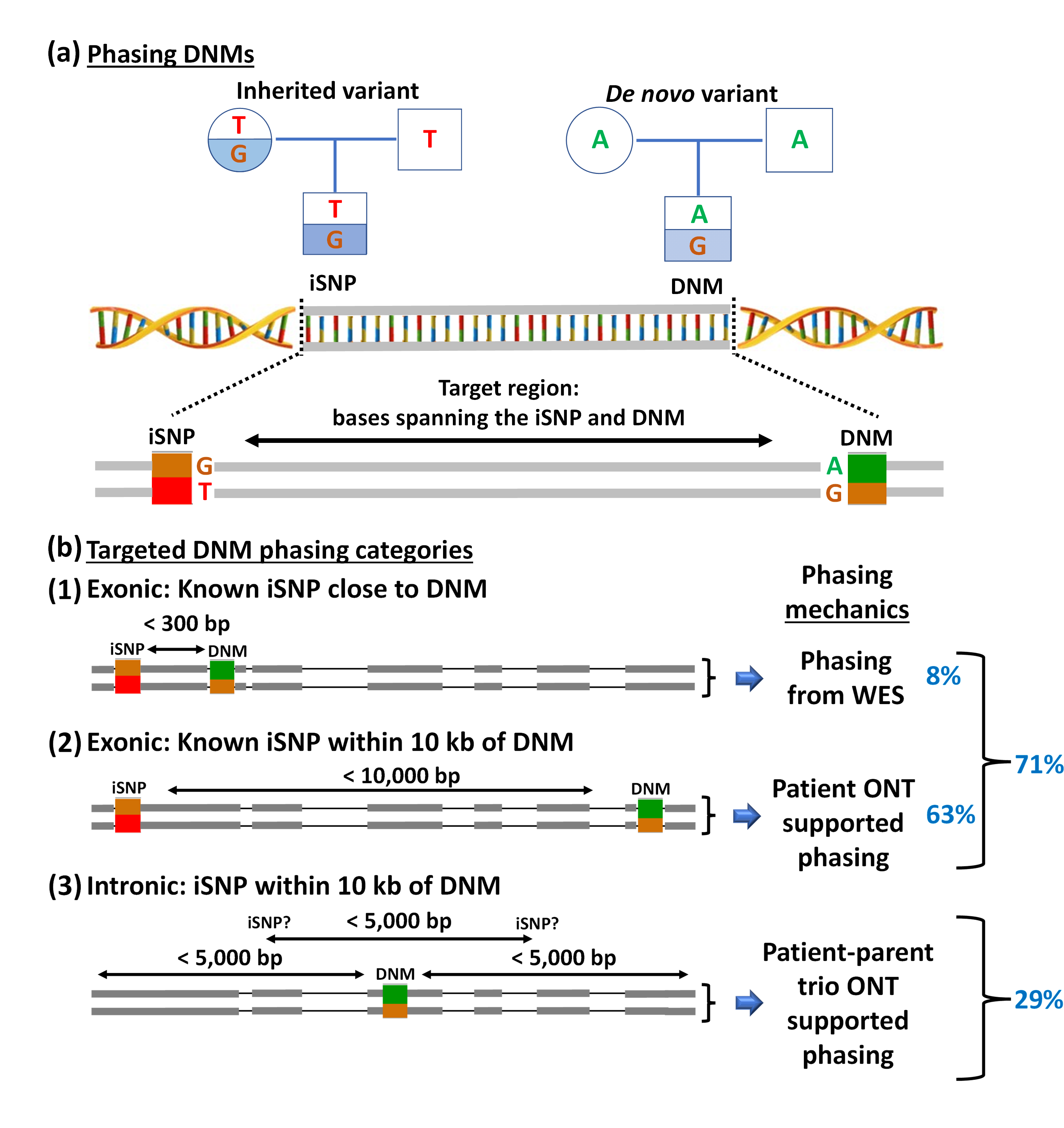
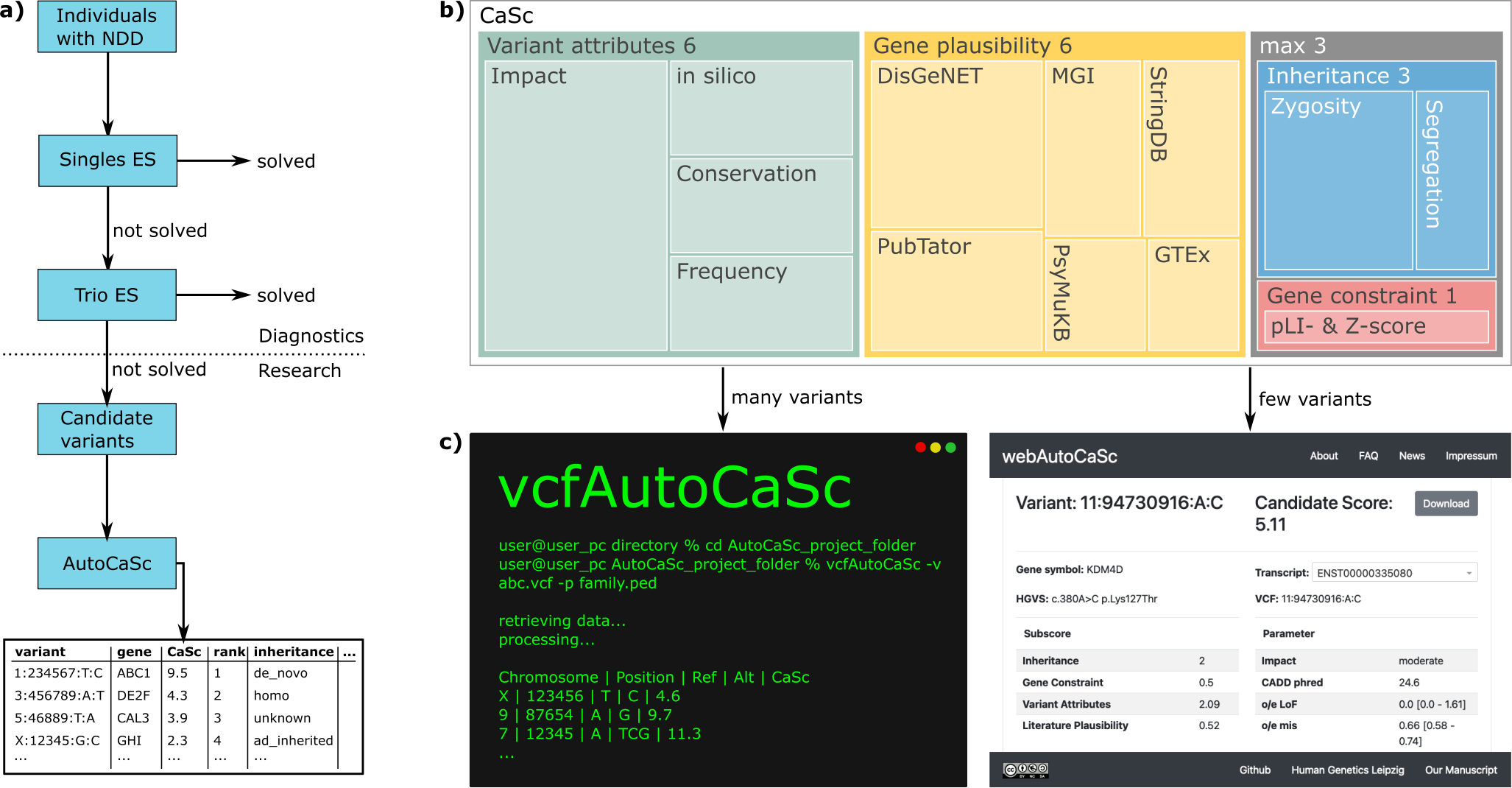
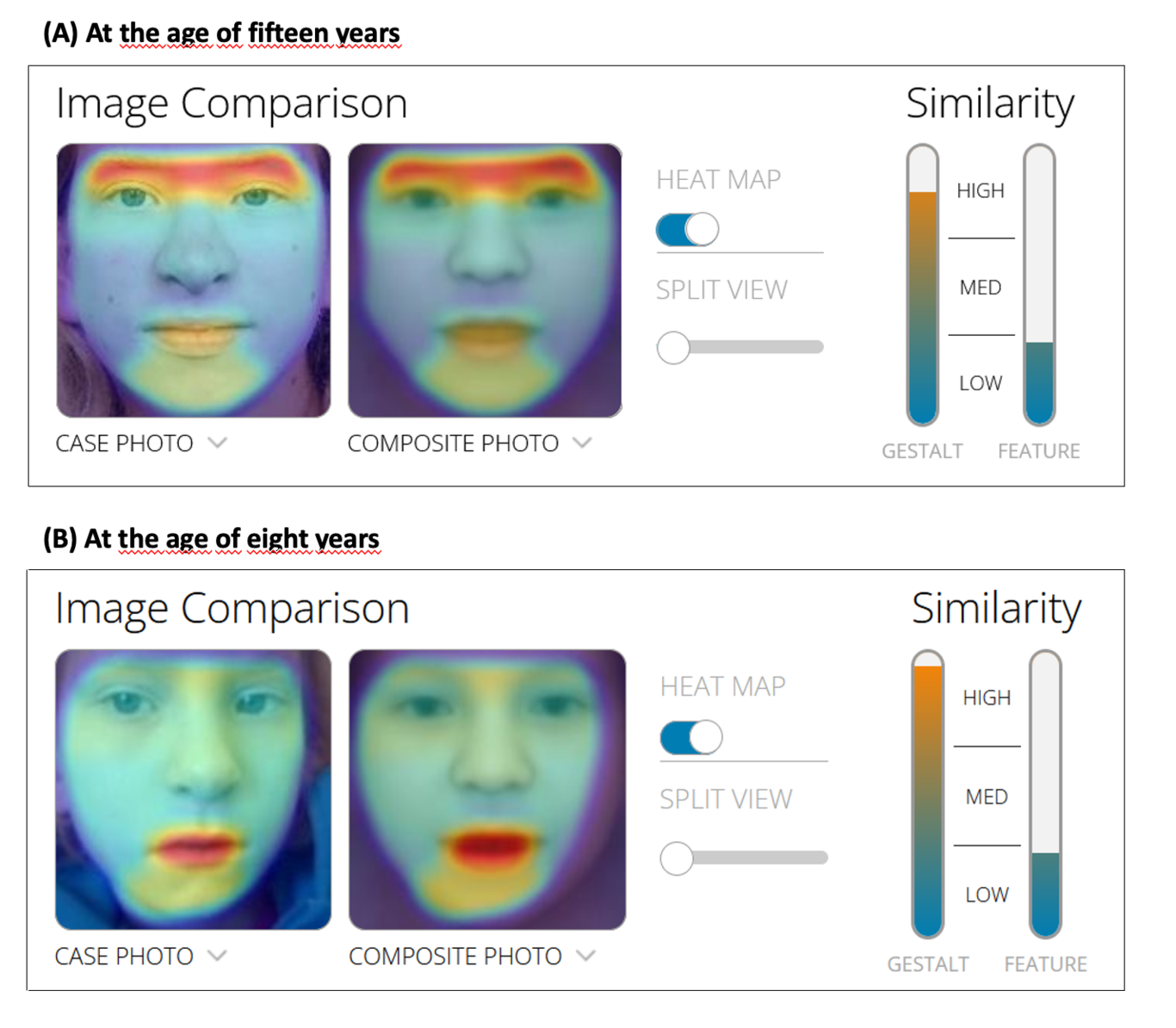
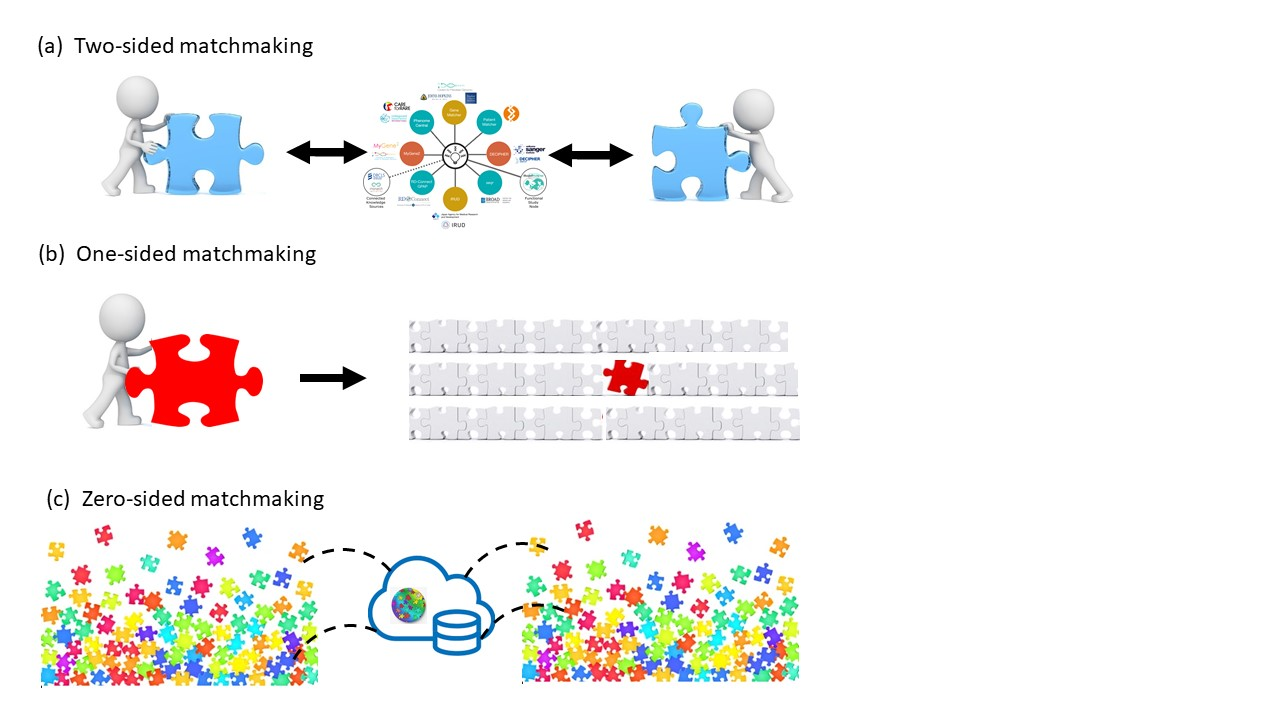
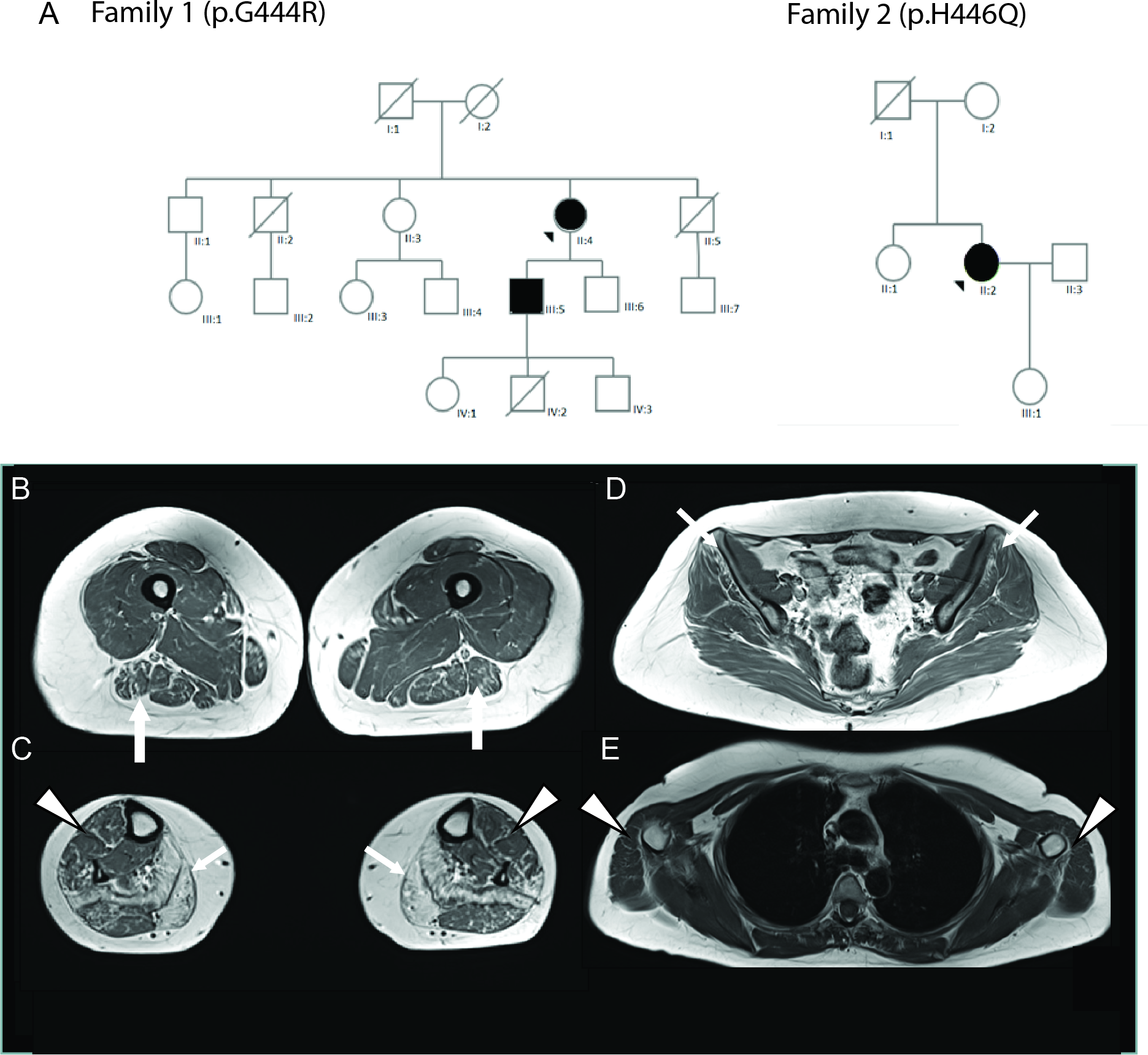
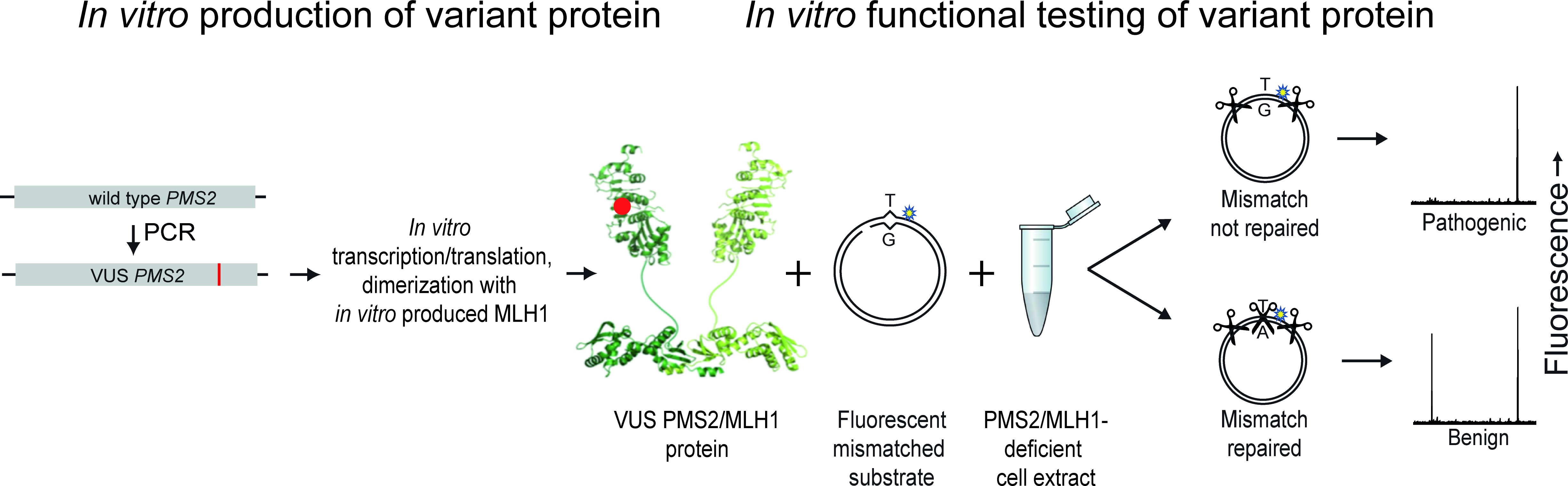
The large majority of germline alterations identified in the DNA mismatch repair (MMR) gene PMS2, a low-penetrance gene for the cancer predisposition Lynch Syndrome (LS, OMIM 120435), represent variants of unknown significance (VUS). The inability to assess pathogenicity of such VUS interferes with personalized healthcare. The complete in vitro MMR activity (CIMRA) assay, that only requires sequence information on the VUS, provides a functional analysis-based tool suited for VUS classification. To derive a formula that translates CIMRA assay results for PMS2 VUS into the odds of pathogenicity (OddsPath), we used a set of clinically classified PMS2 variants, supplemented by inactivating variants generated by an in cellulo genetic screen, as proxies for pathogenic variants. Validation of this OddsPath revealed very high predictive values for PMS2 VUS. We conclude that this OddsPath provides an integral metric that, similar to the other, higher penetrance, MMR proteins MSH2, MLH1 and MSH6, can be incorporated into the upcoming criteria for MMR gene VUS classification of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP). This will represent a seminal step forward in enabling personalized healthcare for individuals suspected of LS and their relatives.