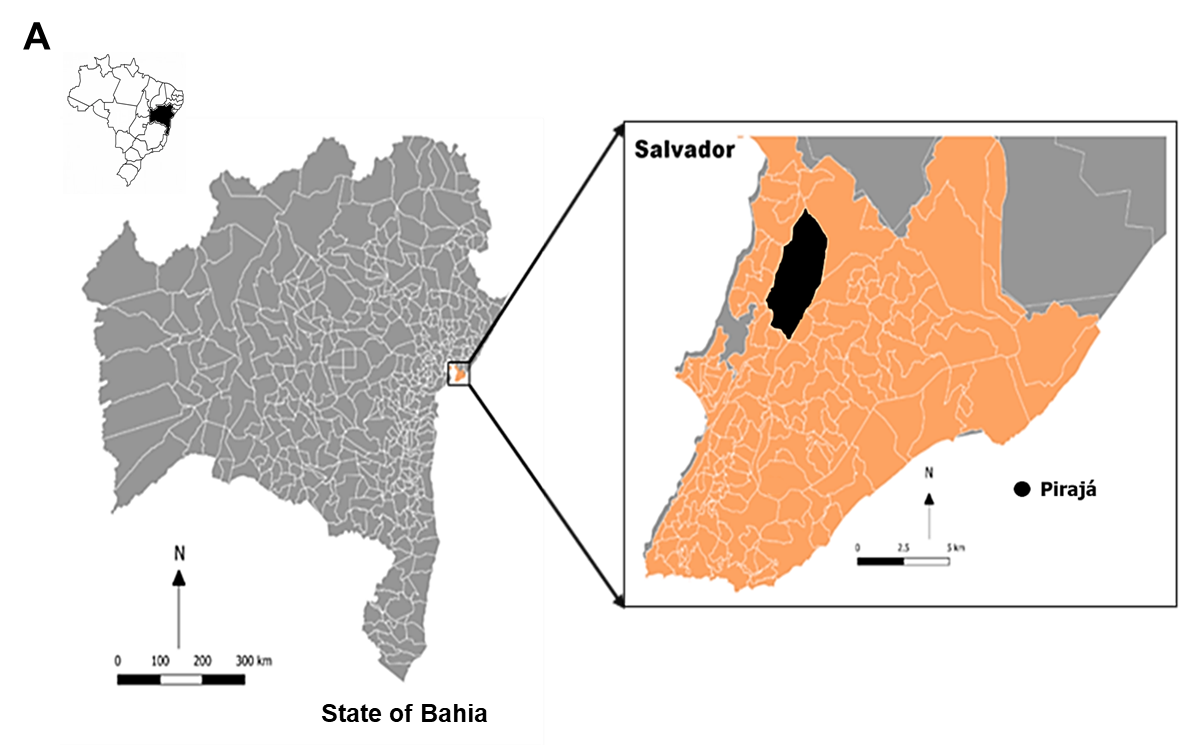
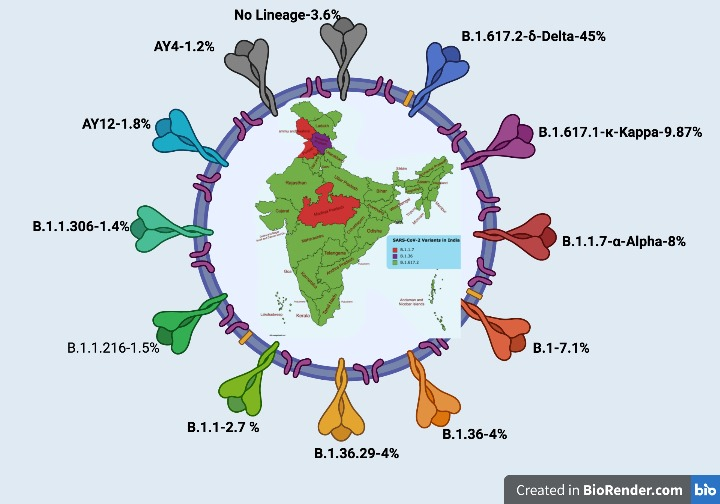
Against the backdrop of the second wave of COVID-19 pandemic in India that started in March 2021, we have monitored the spike (S) protein mutations in all the reported (GISAID portal) whole genome sequences of SARS CoV-2 circulating in India from 1 st January 2021 to 31 st August 2021. In the 43,102 SARS-CoV-2 genomic sequences analysed, we have identified 24, 260 mutations in the S protein, based on which 265 pango lineages could be categorised. The dominant lineage in most of the 28 states of India and its 8 union territories was B.1.617.2 (the delta variant). However, the states Madhya Pradesh, Jammu & Kashmir, and Punjab had B.1.1.7 (alpha variant) as the major lineage, while the Himachal Pradesh state reported B.1.36 as the dominating lineage. A detailed analysis of various domains of S protein was carried out for detecting mutations having a prevalence of >1%; 70, 18, 7, 3, 9, 4, and 1 (N=112) such mutations were observed in the N -terminal domain, receptor binding domain, C -terminal domain, fusion peptide region, heptapeptide repeat (HR)-1 domains, signal peptide domain, and transmembrane region, respectively. However, no mutations were recorded in the HR-2, and cytoplasmic domains of the S protein. Interestingly, 13.39% (N=15) of these mutations were reported to increase the infectivity and pathogenicity of the virus; 2%(N=3) were known to be vaccine breakthrough mutations; and 0.89%(N=1) were known to escape neutralising antibodies. Biological significance of 82% (N=92) of the reported mutations is yet unknown. As SARS-CoV-2 variants are emerging rapidly, it is critical to continuously monitor local viral mutations to understand national trends of virus circulation. This can tremendously help in designing better preventive regimens in the country, and avoid vaccine breakthrough infections.