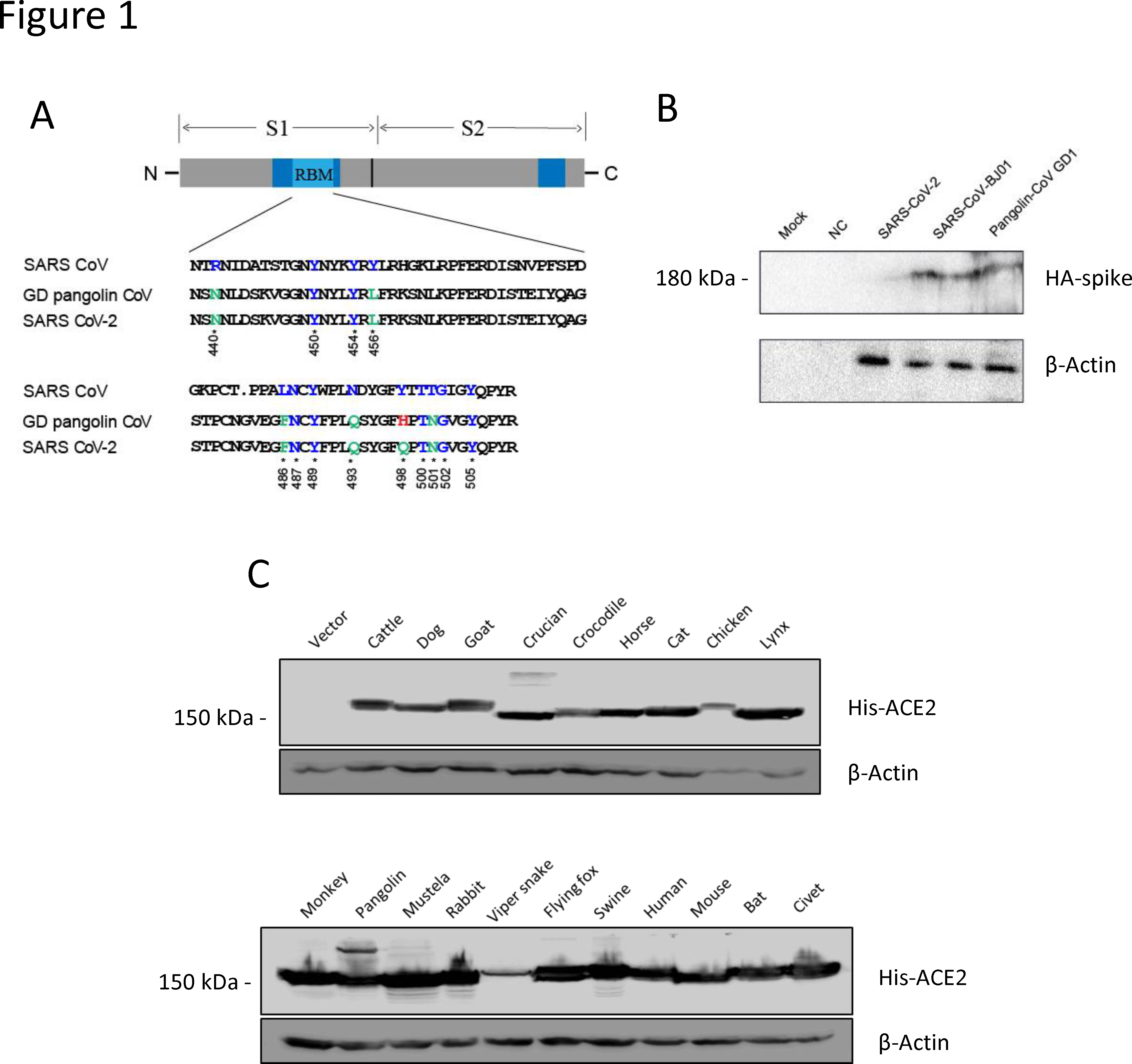
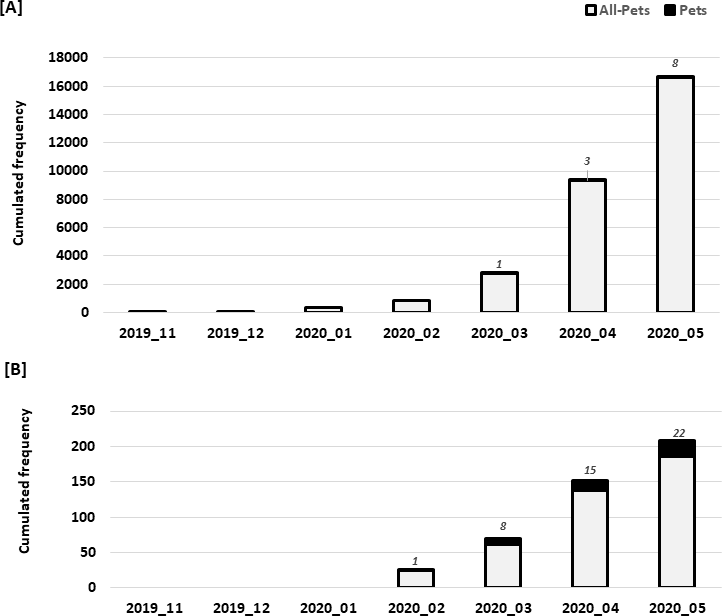
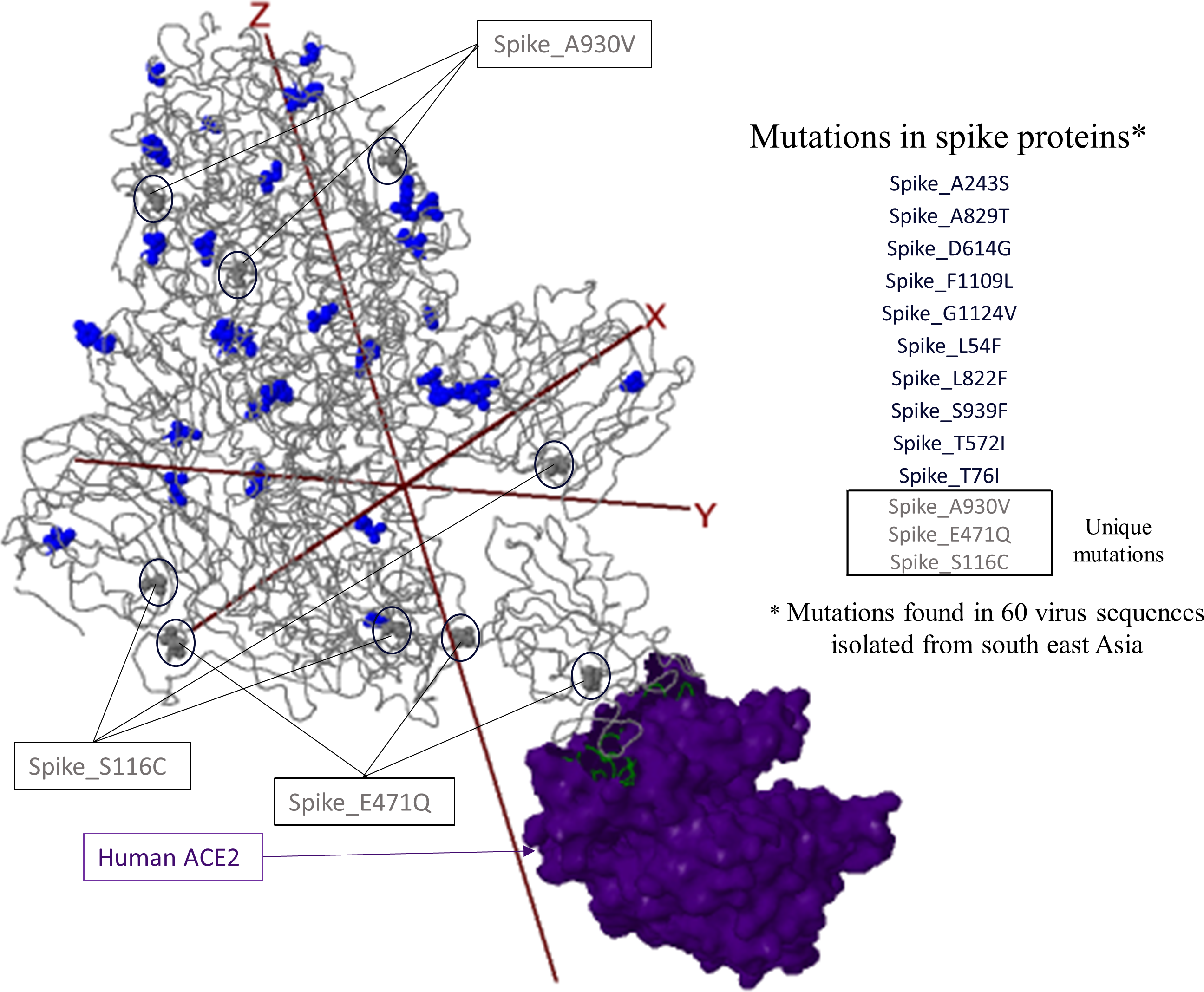
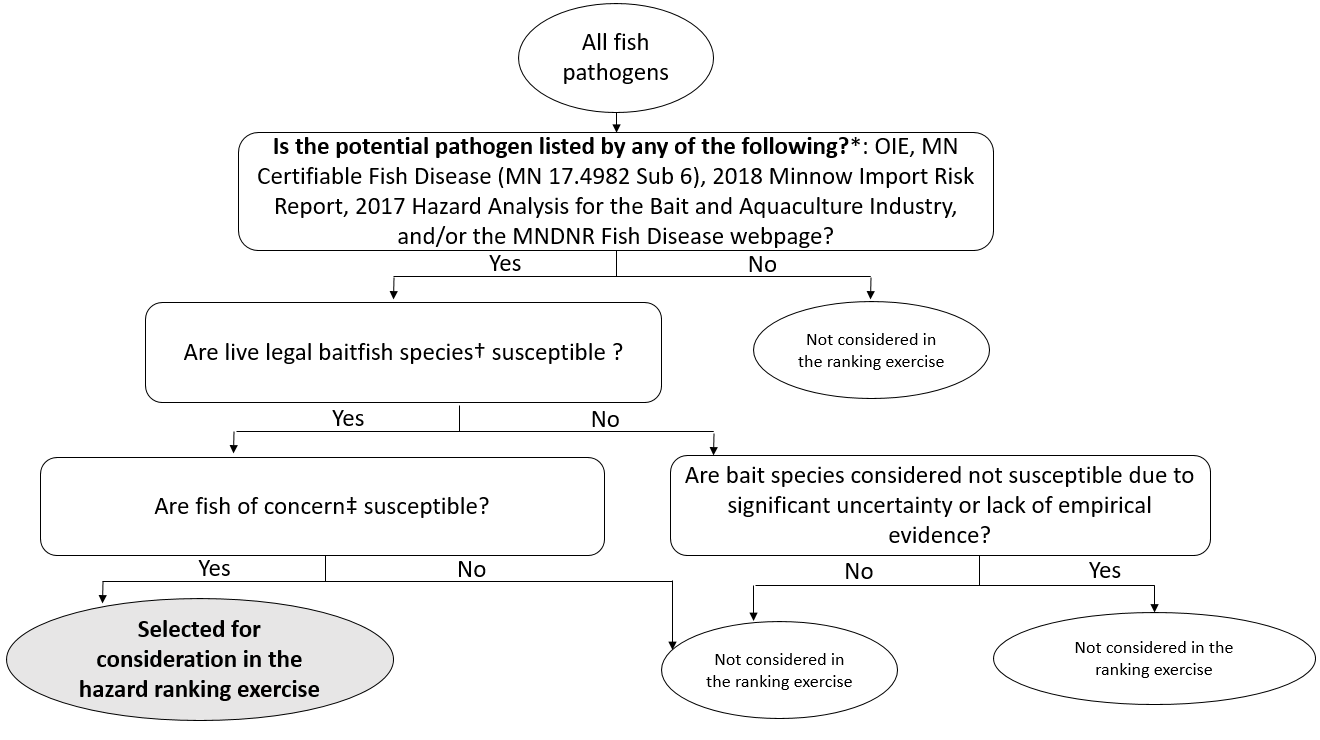
Infection with the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) induces the coronavirus infectious disease 19 (COVID-19). Its pandemic form in human population and its probable animal origin, along with recent case reports in pets, make drivers of emergence crucial in carnivore domestic pets, especially cats, dogs and ferrets. Few data are available in these species; we first listed forty-six possible drivers of emergence of COVID-19 in pets, regrouped in eight domains (i.e. pathogen/disease characteristics, spatial-temporal distance of outbreaks, ability to monitor, disease treatment and control, characteristics of pets, changes in climate conditions, wildlife interface, human activity, and economic and trade activities). Secondly, we developed a scoring system per driver, then elicited experts (N = 33) to: (i) allocate a score to each driver, (ii) weight the drivers scores within each domain and (iii) weight the different domains between them. Thirdly, an overall weighted score per driver was calculated; drivers were ranked in decreasing order. Fourthly, a regression tree analysis was used to group drivers with comparable likelihood to play a role in the emergence of COVID-19 in pets. Finally, the robustness of the expert elicitation was verified. Five drivers were ranked with the highest probability to play a key role in the emergence of COVID-19 in pets: availability and quality of diagnostic tools, human density close to pets, ability of preventive/control measures to avoid the disease introduction or spread in a country (except treatment, vaccination and reservoir(s) control), current species specificity of the disease causing agent and current knowledge on the pathogen. As scientific knowledge on the topic is scarce and still uncertain, expert elicitation of knowledge, in addition with clustering and sensitivity analyses, is of prime importance to prioritize future studies, starting from the top five drivers. The present methodology is applicable to other emerging pet diseases.