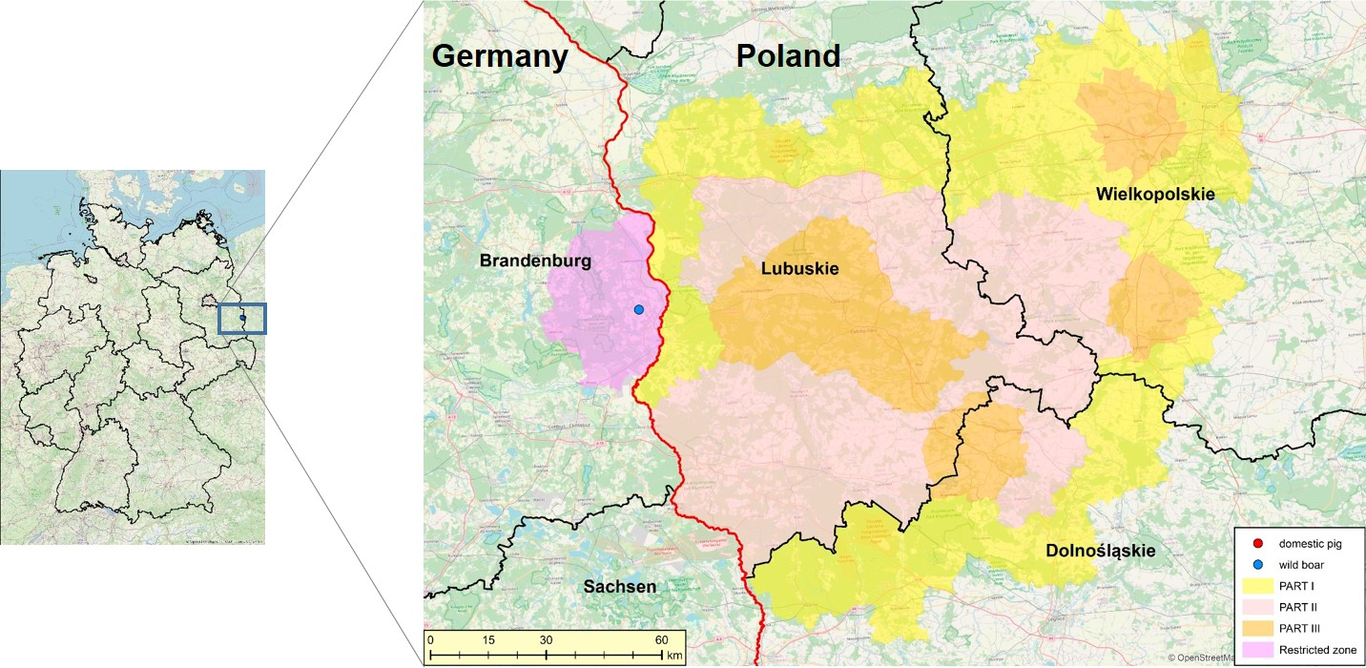
African swine fever (ASF) and classical swine fever (CSF) are two major transboundary animal diseases of swine with important socioeconomic consequences at farm, subnational and national level. The objective of this study was to evaluate the direct cost of outbreaks and their control at country/regional level in four countries: namely CSF in Colombia in 2015-2016, the retrospective cost of ASF in the Philippines in 2019 and in a province of Vietnam in 2020, and a hypothetical ASF scenario in one region in North Macedonia, using the newly developed Outbreak Costing Tool (OutCosT). The tool calculates the costs of 106 different items, broken down by up to four types of farms, and by who assumes the cost (whether veterinary services, farmers or other stakeholders). The total cost of CSF in Colombia was US$ 3.8 million of which 88% represented the cost of the vaccination campaign. For ASF, there were wide differences between countries: US$ 826,911 in Lao Cai (Vietnam), US$ 3,319,666 in North Macedonia and over US$ 58 million in the Philippines. While in the Philippines and Vietnam, 96-98% of the cost occurred in the affected farms, the highest expenditure in North Macedonia scenario was the movement control of the neighbouring and at-risk farms (77%). These important differences between countries depend on the spread of the disease, but also on the production systems affected and the measures applied. Apart from the financial cost, these diseases have other negative impacts, especially in the livelihoods of smallholder farms. The OutCosT tool also allows users to evaluate qualitatively other important aspects related to the epidemics, such as the impact on human health, the environment, animal welfare, socio-economic vulnerability, trading and political response. The main purpose of the OutCosT, which will become a FAO corporate tool, is to support country authorities to rapidly respond to ASF outbreaks by estimating the associated costs, and for advocacy purposes to mobilize resources at national or international levels.