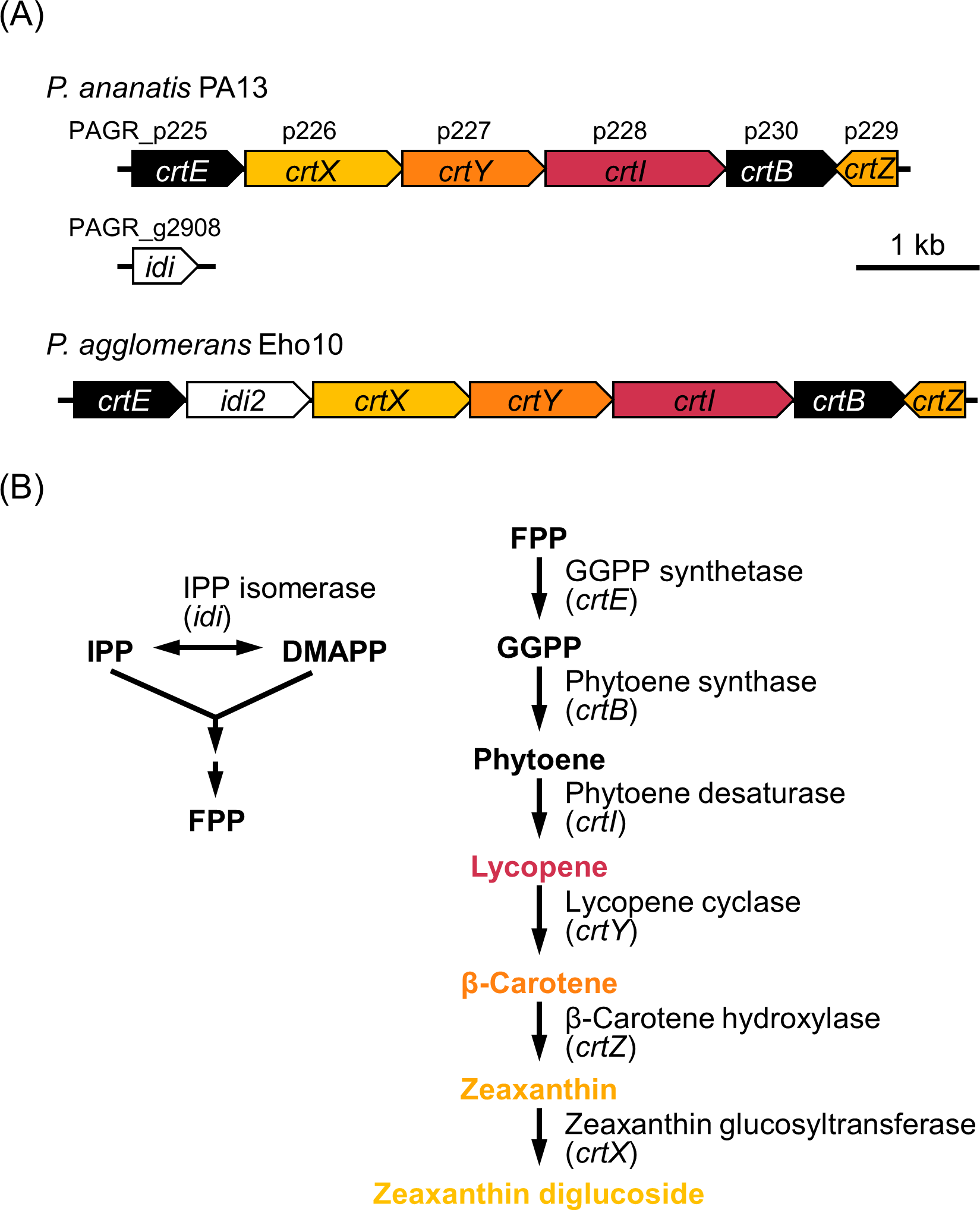
Carotenoids are widely used in functional foods, cosmetics, and health supplements, and their importance and scope of use are continuously expanding. Here, we characterised carotenoid biosynthetic genes of the plant-pathogenic bacterium Pantoea ananatis, which carries a carotenoid biosynthetic gene cluster (including crtE, X, Y, I, B, and Z) on a plasmid. Reverse transcription–polymerase chain reaction (RT-PCR) analysis revealed that the crtEXYIB gene cluster is transcribed as a single transcript and crtZ is independently transcribed in the opposite direction. Using splicing by overlap extension with polymerase chain reaction (SOE by PCR) based on asymmetric amplification, we reassembled crtE–B, crtE–B–I, and crtE–B–I–Y. High-performance liquid chromatography confirmed that Escherichia coli expressing the reassembled crtE–B, crtE–B–I, and crtE–B–I–Y operons produced phytoene, lycopene, and β-carotene, respectively. We found that the carotenoids conferred tolerance to UV radiation and toxoflavin. Pantoea ananatis shares rice environments with the toxoflavin producer Burkholderia glumae and is considered to be the first reported example of producing and using carotenoids to withstand toxoflavin. We confirmed that the carotenoid production of P. ananatis is dependent on RpoS, which is positively regulated by Hfq/ArcZ and negatively regulated by ClpP, similar to an important regulatory network of E. coli (HfqArcZ → RpoS Ͱ ClpXP). We also demonstrated that Hfq-controlled quorum signalling de-represses EanR to activate RpoS, thereby initiating carotenoid production. Survival genes such as those responsible for the production of carotenoids of the plant-pathogenic P. ananatis must be expressed in a timely manner to overcome stressful environments and compete with other microorganisms. This mechanism is likely maintained by a brake with excellent performance, such as EanR.