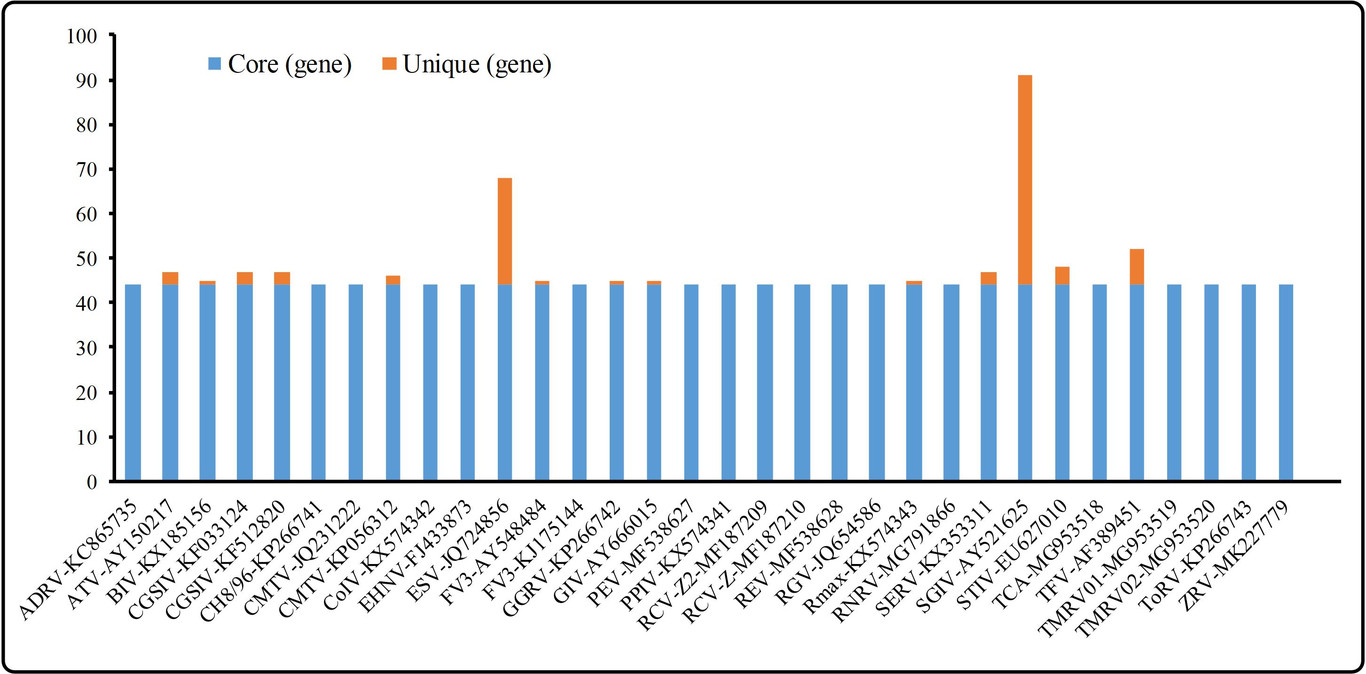
Ranaviruses can infect both captive and wild cold-blooded vertebrates, leading to significant economic and environmental losses. With the cases of ranavirus infection increasing, many ranavirus genomic sequences were published, but little is known about ranavirus taxonomy on a whole genome level. In this study, 44 ranaviruses core genes were identified in 32 ranaviruses genome suquences by using PanX. The Neighbor joining phylogenetic trees (NJ-tree) based on 44 ranaviruses core genes and 24 iridoviridae core genes and composition vector phylogenetic tree (CV-Tree) based on whole genome were constructed. The three of phylogenetic trees showed that 32 ranavirus isolates can be divided to 4 different subspecies including GIV-like, EHNV-like, FV3-like and CMTV-like, and subspecies taxonomic position of three phylogenetic trees were consistent. However, the phylogenetic position of ToRV could not be determined if it belongs to FV3-like or CMTV-like group. Subsequently, we carried out dot plot analysis and confirmed that ToRV should belong to CMTV-like group. Based on dot plot analysis and phylogenetic trees, taxonomic classification of ranaviruses were confirmed. Finally, 4 genes which are suitable for the construction of phylogenetic tree were selected from ranavirus core genes by recombination analysis, substitution saturation analysis and single-gene phylogenetic analysis. Phylogenetic tree based on concatenated sequences of the 4 selected genes showed that classification of subspecies was identical with 3 of the phylogenetic trees. Conclusion: our results confirmed taxonomic identification of ranaviruses, the 4 selected genes used in phylogenic analysis will make taxonomic identification more convenient and accurate.