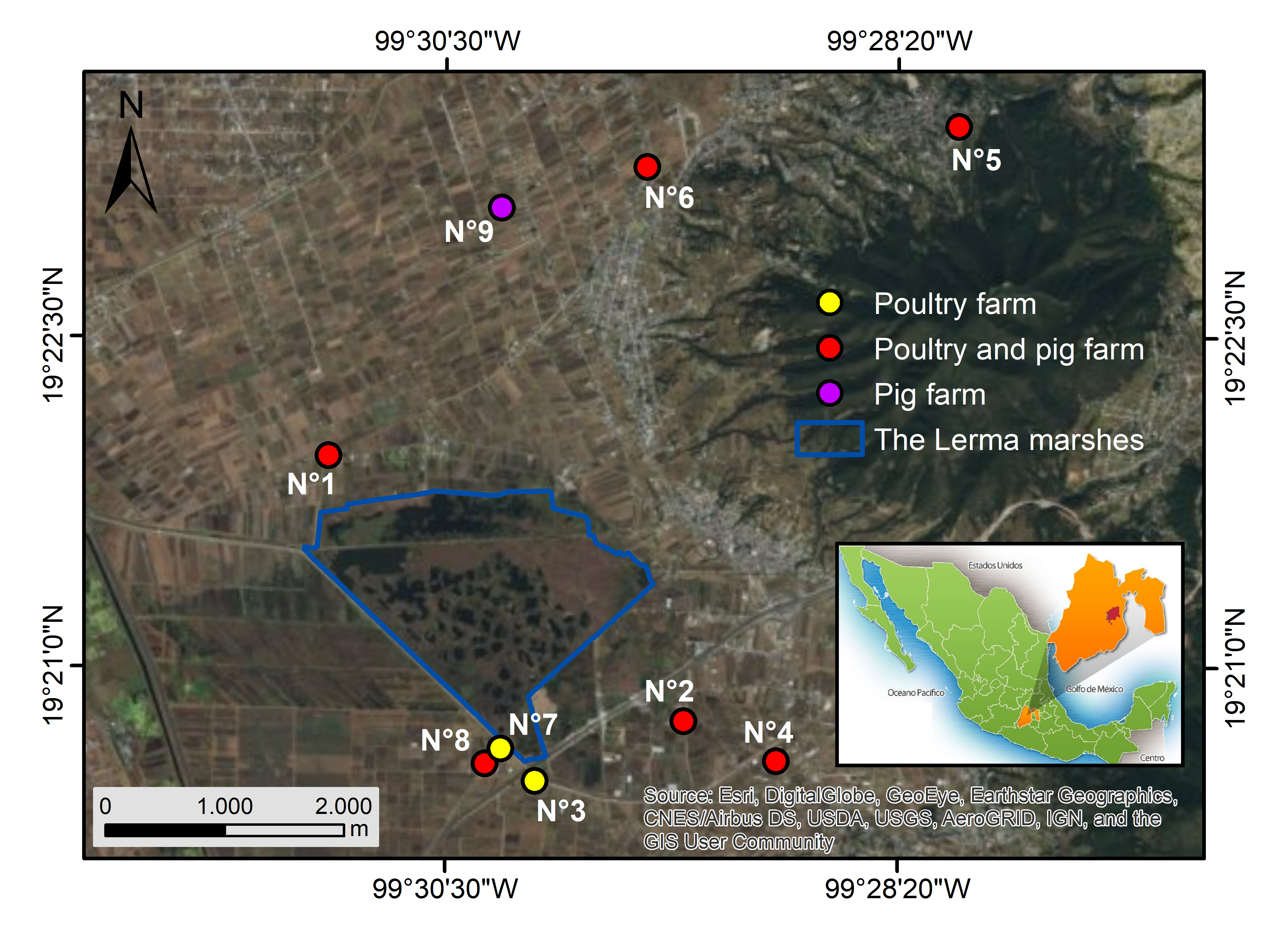
Influenza A virus (IAV) outbreaks constitute a constant threat to public health and pose a remarkable impact on socio-economic systems worldwide. Interactions between wild and domestic birds, humans, and swine can lead to spillover events. Backyard livestock systems in proximity to wetlands represent a high-risk area for viral spread. However, some gaps remain in our knowledge of IAV transmission at the wildlife – livestock interface in Mexico. Hence, the study aimed at molecular identification and phylogenetic characterization of IAV in the wild duck – backyard livestock interface at a wetland of Mexico. A total of 875 animals were tested by real-time RT-PCR (qRT-PCR). We detected IAV in 3.68% of the wild ducks sampled during the winter season 2016 – 2017. Nonetheless, the samples obtained from backyard poultry and swine tested negative. The highest IAV frequency (11.10%) was found in the Mexican duck (Anas diazi). Subtypes H1N1, H3N2, and H5N2 were detected. Phylogenetic analysis of influenza viruses isolated from wild ducks of the Lerma marshes revealed that hemagglutinin (HA) gene sequences were related to waterfowl, swine, and poultry IAV strains previously isolated in the United States and Mexico. In conclusion, the co-circulation of three IAV subtypes in wild ducks close to backyard farms in Mexico, as well as, the local identification of HA gene sequences genetically related to Mexican livestock IAV strains and also to North American waterfowl IAV strains, highlight the importance of the Lerma marshes for influenza surveillance given the close interaction among wild birds, poultry, pigs, and humans.