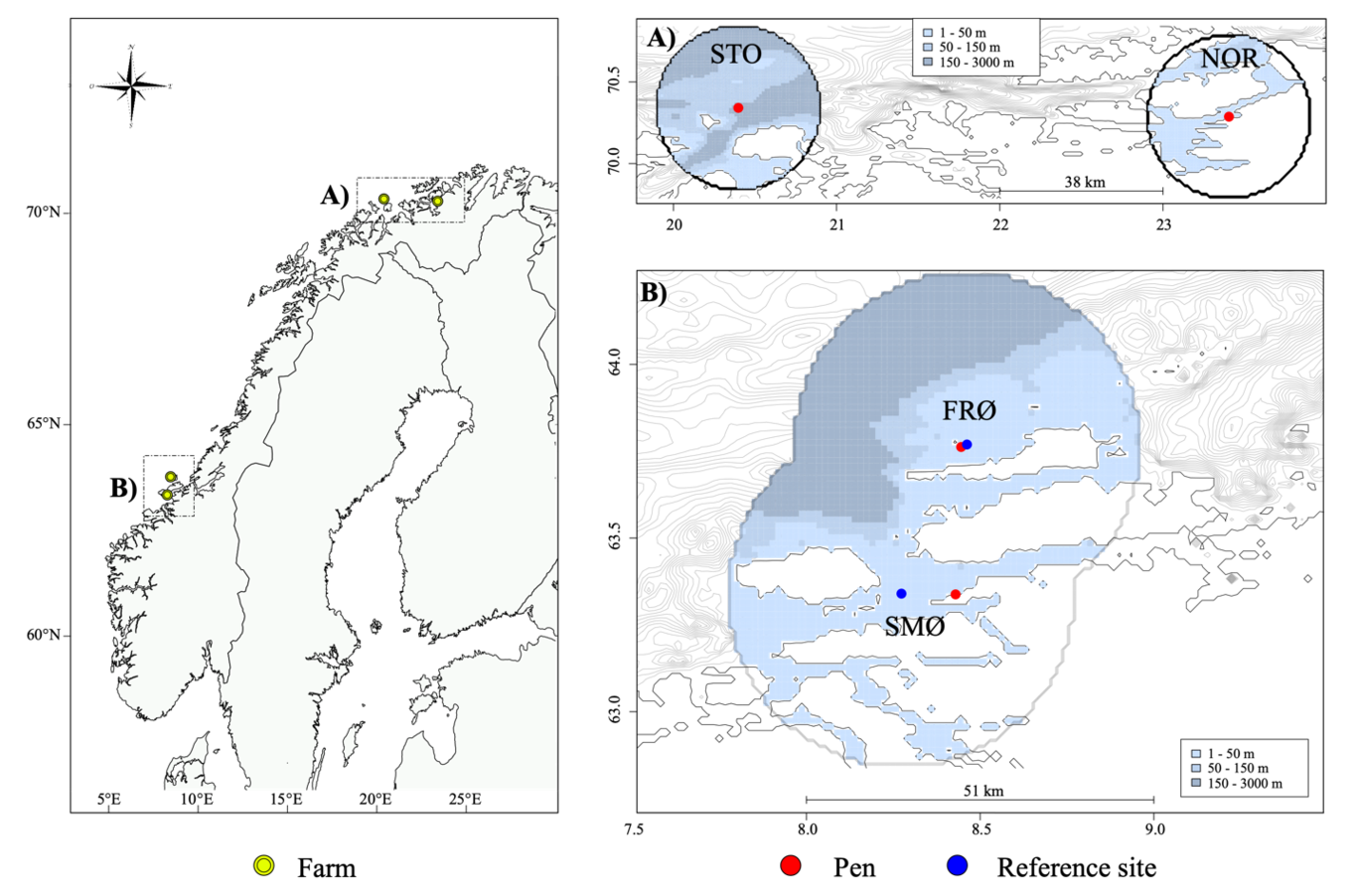
Molecular biosecurity surveillance programs increasingly use environmental DNA (eDNA) for detecting marine non-indigenous species (NIS). However, the current molecular detection workflow is cumbersome, prone to errors and delays, and is limited in providing knowledge about eDNA beyond the spatial and temporal extent of the sampling. These limitations can hinder management efforts and restrict the “opportunity window” for a rapid response to new marine NIS incursions. Emerging innovative field-deployable digital droplet PCR (ddPCR) systems offer improved workflow efficiency by autonomously analyzing targeted free-floating extra-cellular eDNA (free-eDNA) signals. Despite their potential, these systems have not been tested in marine environments. Thus, an aquarium study was conducted with three distinct marine NIS: the Mediterranean fanworm Sabella spallanzanii, the ascidian clubbed tunicate Styela clava, and the brown bryozoan Bugula neritina to evaluate the detectability of free-eDNA in seawater. The detectability of targeted free-eDNA was assessed by directly analyzing aquarium water samples using an optimized species-specific ddPCR assay, without filtration or DNA extraction, so-called, “direct-ddPCR”. The results demonstrated the consistent detection of Sabella spallanzanii and Bugula neritina free-eDNA when these organisms were present in high abundance. Once organisms were removed, the free-eDNA signal exponentially declined, noting that free-eDNA persisted between 24-72 hours. Results indicate that organism biomass, specimen characteristics (e.g., stress and viability), and species-specific biological differences may influence free-eDNA detectability. These results are critical for implementing in-situ nucleic acid automated continuous sensing systems for marine biosurveillance, enabling point-of-need detection and rapid management response to biosecurity threats.