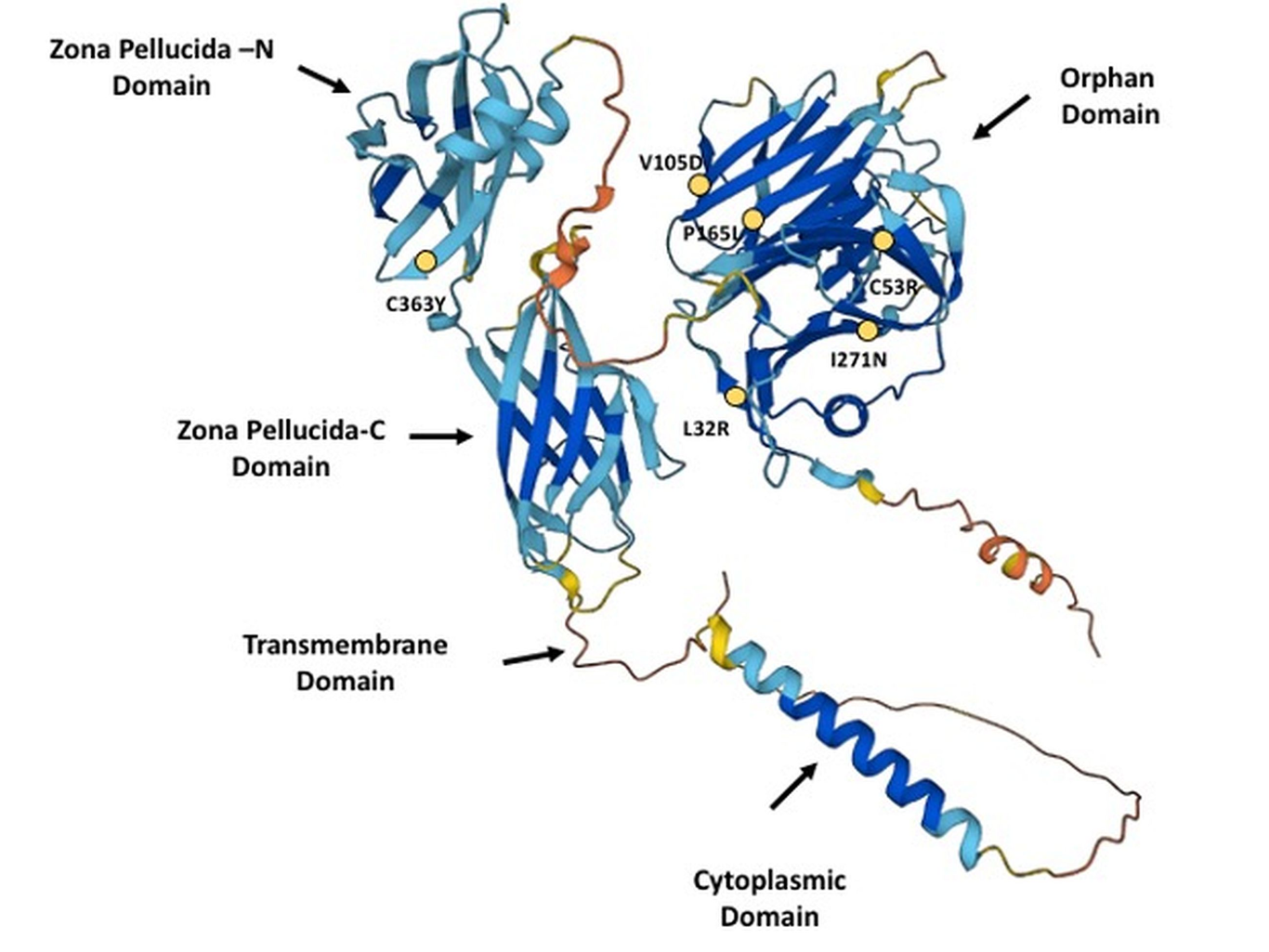
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder affecting 1 in 5,000-8,000 individuals. Hereditary hemorrhagic telangiectasia type 1 (HHT1) is the most common HHT and manifests as diverse vascular malformations ranging from mild symptoms such as epistaxis and mucosal and cutaneous telangiectases to severe arteriovenous malformations (AVMs) in the lungs, brain or liver. HHT1 is caused by heterozygous mutations in the ENG gene, which encodes endoglin, the TGFb homodimeric coreceptor. It was previously shown that some endoglin HHT1-causing variants failed to traffic to the plasma membrane due to their retention in the endoplasmic reticulum (ER) and consequent degradation by ER-associated degradation (ERAD). Endoglin is a homodimer formed in the ER, and we therefore hypothesized that mixed heterodimers might form between ER-retained variants and WT protein, thus hampering its maturation and trafficking to the plasma membrane causing dominant negative effects. Indeed, HA-tagged ER-retained mutants formed heterodimers with Myc-tagged WT Endoglin. Moreover, variants L32R, V105D, P165L, I271N and C363Y adversely affected the trafficking of WT endoglin by reducing its maturation and plasma membrane localization. These results strongly suggest dominant negative effects exerted by these ER-retained variants aggravating endoglin loss of function in patients expressing them in the heterozygous state with the WT allele. Moreover, this study may explain some of the variability observed among HHT1 patients due to the additional loss of function exerted by the dominant negative effects in addition to that due to haploinsufficiency. These findings might also have implications for some of the many conditions impacted by ERAD.