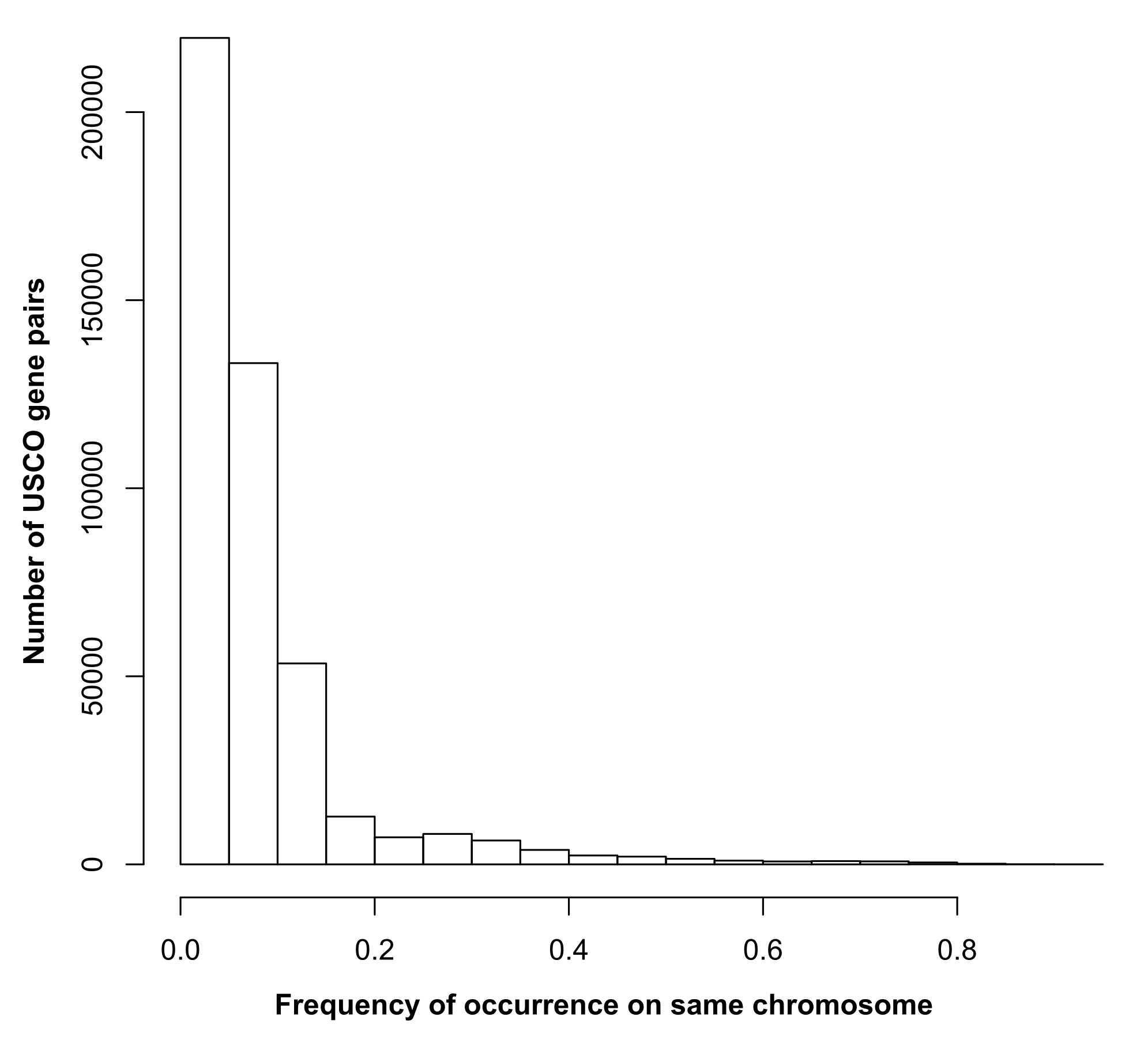
Spiders are a hyper-diverse taxon and among the most abundant predators in nearly all terrestrial habitats. Their success is often attributed to key developments in their evolution such as silk and venom production and major apomorphies such as a whole-genome duplication. Resolving deep relationships within the spider tree of life has been historically challenging, making it difficult to measure the relative importance of these novelties. Whole-genome data offer an essential resource in these efforts, but also for functional genomic studies. Here, we present de novo assemblies for three spider species: Ryuthela nishihirai (Heptathelidae), a representative of the ancient Mesothelae, the suborder which is sister to all other extant spiders; Uloborus plumipes (Uloboridae), a cribellate orbweaver whose phylogenetic placement is especially challenging; and Cheiracanthium punctorium (Cheiracanthiidae), which represents only the second family to be sequenced in the hyper-diverse Dionycha clade. These genomes fill critical gaps in the spider tree of life. Using these novel genomes along with 25 previously published ones, we examine two proposed drivers of diversification: spidroin gene and structural hox cluster diversity. Our analyses show that spidroin diversification as well as hox cluster duplication and restructuring mirror spider diversification, hence suggesting a key role in the evolutionary success of the group. Our assemblies provide critical genomic resources to facilitate deeper investigations into spider evolution. The near chromosome-level genome of the “living fossil” R. nishihirai represents an especially important step forward, offering new insights into the origins of spider traits.