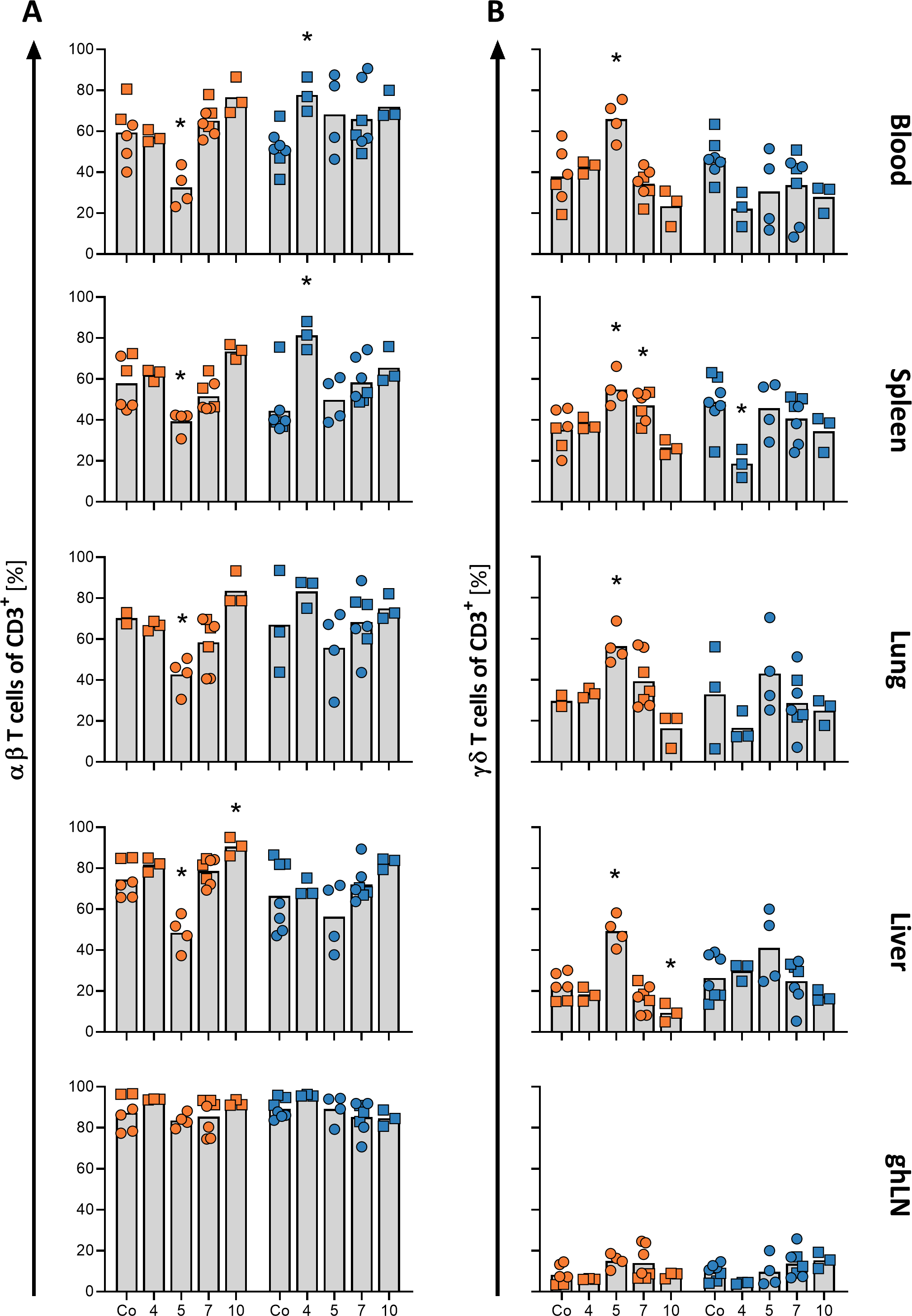
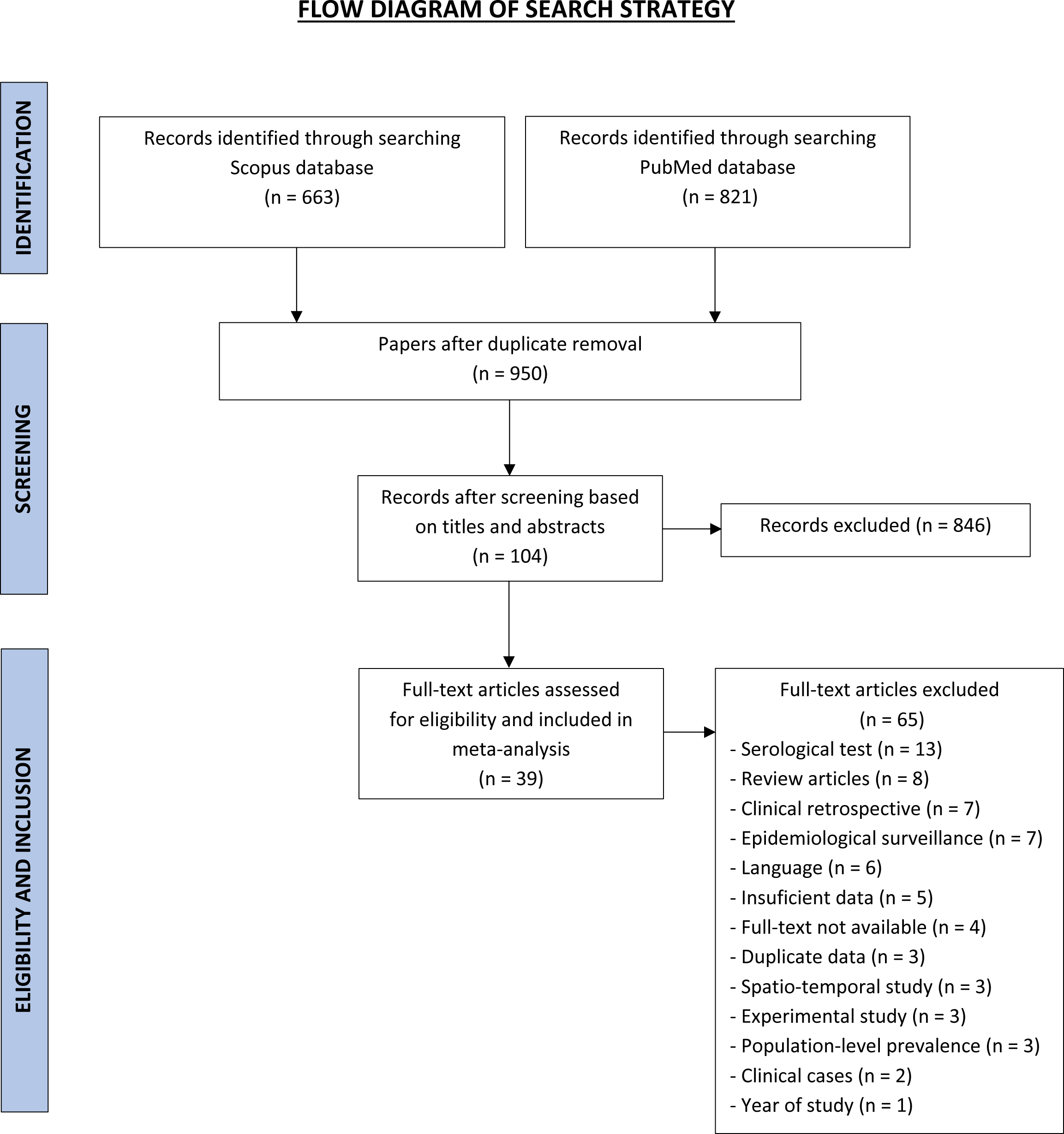
African swine fever virus (ASFV) can survive in soy-based products for 30 days with T ½ ranging from 9.6-12.9 days in soybean meals and soy oil cake. As the US imports soy-based products from several ASFV-positive countries, knowledge of the type and quantity of these specific imports, and their ports of entry (POE), is necessary information to manage risk. Using the data from the International Trade Commission Harmonized Tariff Schedule website in conjunction with pivot tables, we analyzed imports across air, land, and sea POE of soy-based products from 43 ASFV-positive countries to the US during 2018 and 2019. In 2018, 104,366 metric tons (MT) of soy-based products, specifically conventional and organic soybean meal, soybeans, soy oil cake and soy oil were imported from these countries into the US via seaports only. The two largest suppliers were China (52.7 %, 55,034 MT) and the Ukraine (42.9%, 44,775 MT). In 2019, 73,331 MT entered the US and 54.7% (40,143 MT) came from the Ukraine and 8.4% (6,182 MT) from China. Regarding POE, 80.9% to 83.2% of soy-based imports from China entered the US at the seaports of San Francisco, CA and Seattle, WA, while 89.4% to 100% entered from the Ukraine via the seaports of New Orleans, LA and Charlotte, NC. Analysis of five-year trends (2015 to 2019) of the volume of soy imports from China indicated reduction over time (with a noticeably sharp decrease between 2018 and 2019), and seaport utilization was consistent. In contrast, volume remained high for Ukrainian soy imports, and seaport utilization was inconsistent. Overall, this exercise introduced a new approach to collect objective data on an important risk factor, providing researchers, government officials, and industry stakeholders a means to objectively identify and quantify potential channels of foreign animal disease entry into the US.